Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
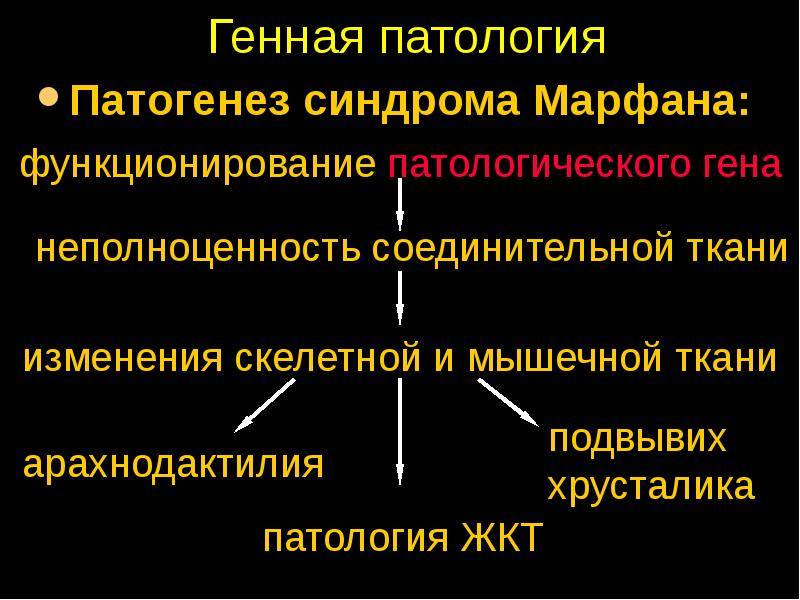
Патогенез синдрома Марфaна
Исследования показали, что происходит мутирование гена белка фибриллина в хромосоме, но с чем это связано, до конца не выявлено. 75% случаев болезни мутированный ген переходит от родителей к детям, в остальных случаях происходят изменения в сперматозоиде или яйцеклетке в момент зачатия, этот процесс может наблюдаться даже у здоровых родителей.
Клиника синдрома Марфaна
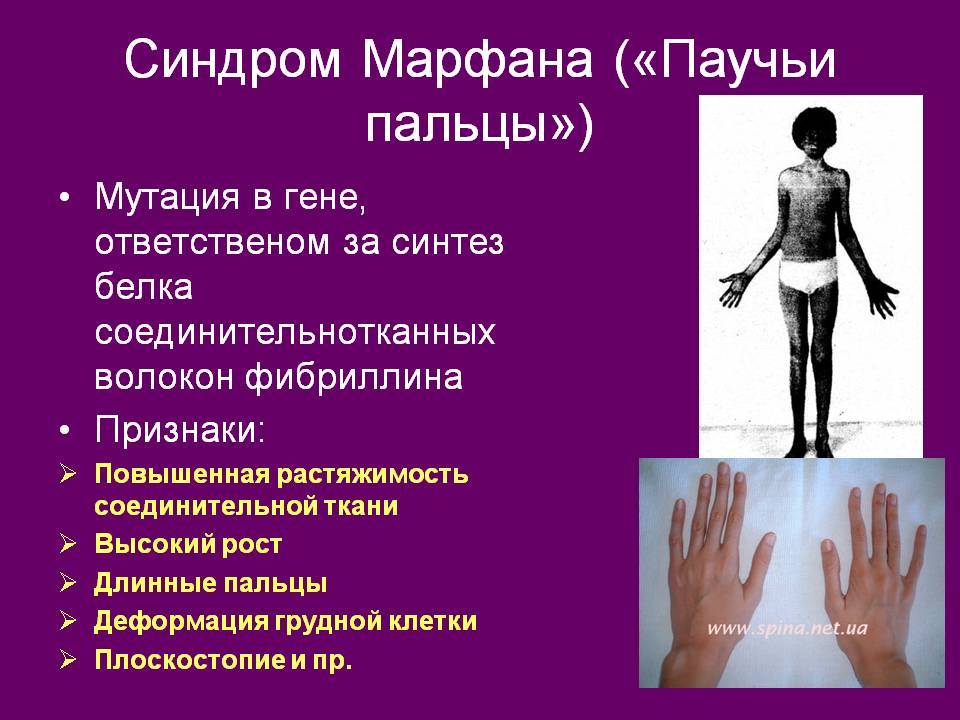
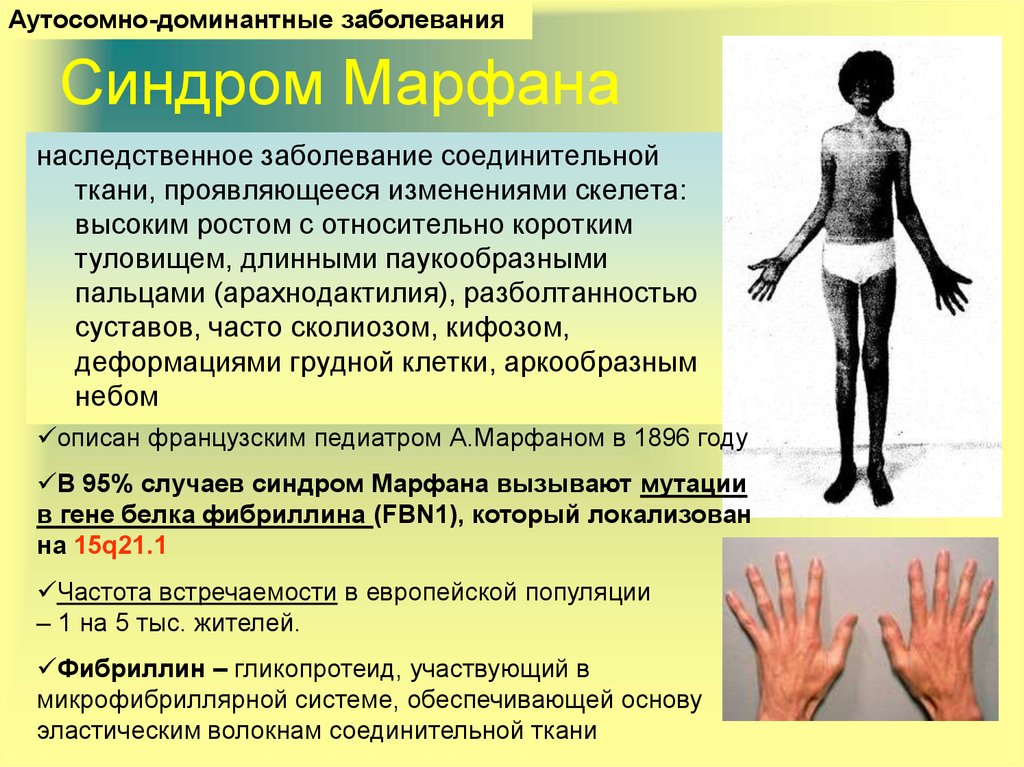
Внешне люди с синдромом Марфина обращают на себя внимание высоким ростом, астеническим телосложением, длинными (непропорционально длинными) конечностями с большим размахом рук, с длинными и очень тонкими пальцами. У них глубоко посаженные глаза, маленькая челюсть, удлиненный череп, высокое нёбо и неправильный прикус
Грудная клетка деформирована («грудь сапожника» или «куриная грудь»), выражен сколиоз, сутулость, часто пациенты предъявляют жалобы на плоскостопие, пальцы стопы молоткообразные. Из-за высокого нёба и маленьких челюстей пациенты невнятно разговаривают. Больные также предъявляют жалобы на боль в суставах и мышцах.
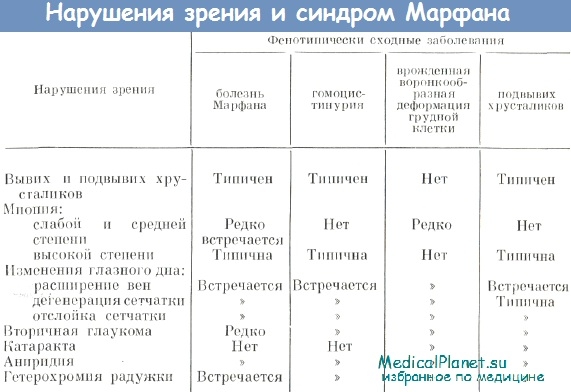
Так же наблюдаются изменения со стороны глаз. Часто у больных отслаивается сетчатка, появляется катаракта, глаукома, у 50% больных с Marfan-Syndrom – «вывих хрусталика» и близорукость. Иногда наблюдается астигматизм и дальнозоркость.
Серьезные изменения у таких больных происходит с сердечно-сосудистой системой. Корень аорты расслаивается и расширяется, возникает аневризма аорты, при разрыве аорты возможен летальный исход. Появляется аритмия, шум в сердце из-за проблем с сердечным клапаном. Формируются пороки сердца, пациентов беспокоят приступы стенокардии.
Болезнь Марфина влияет и на нервную систему. Растягивается твердая оболочка мозга, в итоге появляется боль в пояснице, в области живота, в ногах, головная боль. Эти симптомы исчезают, когда пациент ложится на твердую ровную поверхность.
Может возникнуть спонтанный пневмоторакс. У больного появляется резкая боль в груди, выраженная одышка, цианоз, бледность. Возможна остановка дыхания во сне.
Диагностика Marfan-Syndrom
- Анамнез с указанием этого заболевания у родственников.
- Наличие сочетания таких признаков как аневризма аорты, длинные конечности, дислокация хрусталика, дает основание поставить диагноз Marfan-Syndrom.
- Эхокардиография.
- Электрокардиограмма.
- Осмотр глаз с помощью щелевой лампы.
Лечение Marfan-Syndrom
Лечение симптоматическое, то есть лечение осложнений. Специфического лечения пока нет. Marfan-Syndrom лечится по мере своего развития. При сколиозе возможно ношение бандажа, хирургическая коррекция, физиотерапевтические процедуры.
Прогноз Marfan-Syndrom
Серьезный прогноз при вовлечении в процесс органов сердечно-сосудистой системы. Исход может быть летальным при разрыве аорты.
Причины
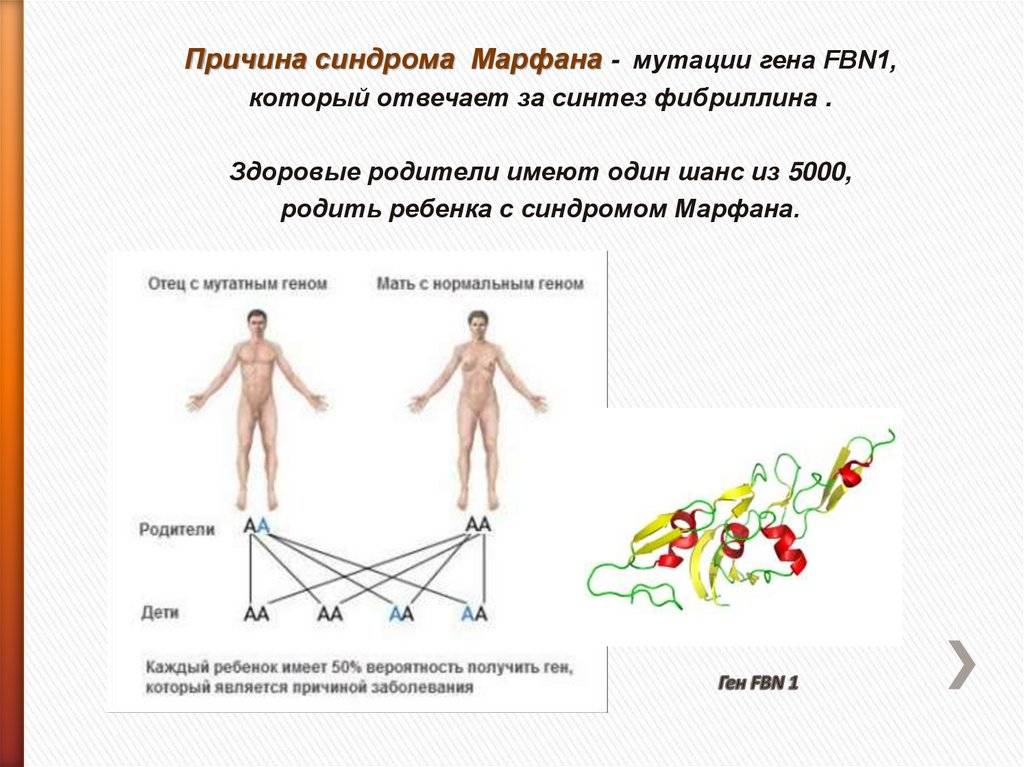
Синдром развивается в результате мутации гена, кодирующего биосинтез особого белка фибриллина — важной структуры межклеточного вещества, которая обеспечивает эластичность и сократительную способность соединительнотканных волокон. Мутация происходит спонтанно в момент зачатия в яйцеклетке или сперматозоиде
При недостатке фибриллина нарушается процесс формирования волокон. Он перестают быть прочными и упругими, становятся чрезмерно растяжимыми и менее устойчивыми к деформациям. В наибольшей степени повреждению подвержены сосуды и связки.
Фибриллин необходим для работы цинновой связки, с помощью которой хрусталик прикрепляется к ресничному телу. При дефиците белка эта связка ослабляется, что проявляется миопией, подвывихом хрусталика, вторичной глаукомой, снижением остроты зрения. Кроме зрительного анализатора, белок фибриллин содержится в связках аорты и обеспечивает ее устойчивость к нагрузкам. При ослаблении этих связок происходит расширение сосуда и расслоение его стенок. Подобные изменения являются смертельно опасными для человека. При синдроме Марфана часто отмечается поражение двустворчатого клапана, что требует проведения хирургической коррекции.
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста – TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных – первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).
Классификация синдрома
Формы патологии:
- стертая – у больных имеются незначительные изменения в 1 или 2 системах организма;
- выраженная – наличие слабовыраженных нарушений в 3 системах или характерных патологических расстройств хотя бы в 1-ой системе.
Характер течения синдрома:
- прогрессирующий – с течением времени патология нарастает и усугубляется,
- стабильный – признаки болезни на протяжении многолетних наблюдений остаются неизменными.
Этиологическая классификация:
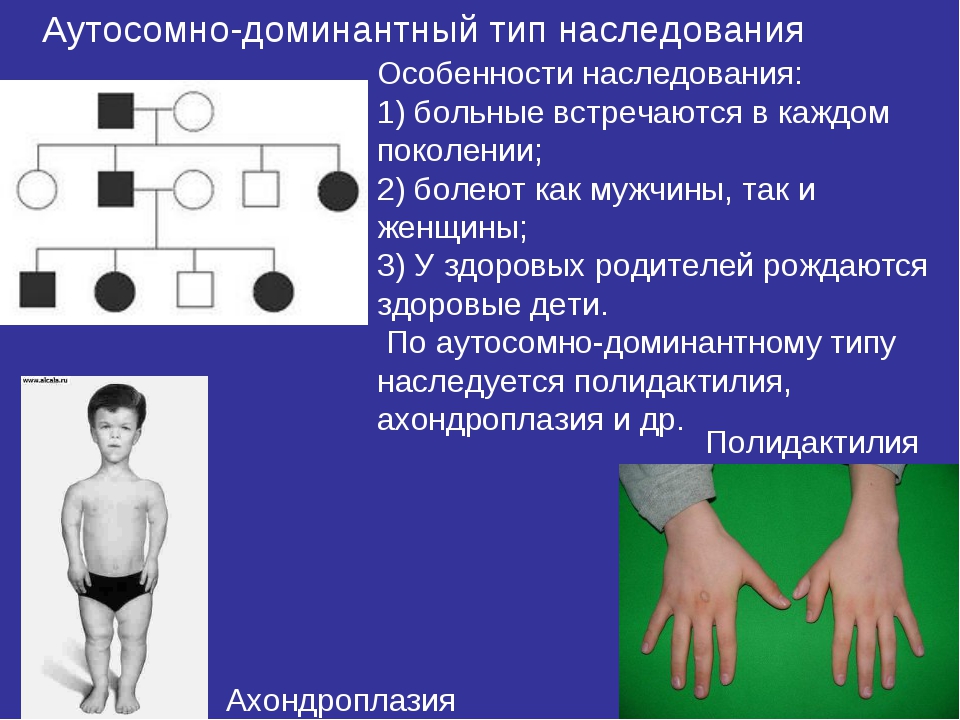
- семейная форма — наследуется по аутосомно-доминантному принципу;
- спорадическая форма — синдром обусловлен случайной мутацией генов во время зачатия.
Лечение
Лечение синдрома Марфана всегда требует одновременного участия нескольких специалистов: кардиолога, кардиохирурга, офтальмолога, ортопеда-травматолога, терапевта.
Спектр лечебных процедур охватывает консервативные и оперативные способы лечения. Консервативные меры направлены на профилактику осложнений и поддержание нормального функционирования органов и систем, а оперативные предполагают коррекцию имеющихся анатомических изменений с целью предотвратить выраженное нарушение функций или даже угрозу для жизни больного.







Хирургические методы лечения:
- реконструктивные операции на аорте (при значительном, более 5 см, расширении восходящей части аорты и расслоении ее стенки);
- протезирование клапанов сердца;
- удаление измененного хрусталика с заменой его на искусственный;
- пластика позвоночника при выраженном сколиозе;
- эндопротезирование тазобедренных суставов;
- пластика грудной клетки (в последние годы отрицается ее целесообразность).
Больному необходим подбор очков или контактных линз, иногда возможна лазерная коррекция зрения.
Медикаментозное лечение имеет патогенетическую и симптоматическую направленность. Больному с метаболической целью назначают большие дозы витамина С (1-3 г в сутки), препараты с глюкозаминосульфатами и хондроитинсульфатами (Терафлекс, Структум, Хондроксид, Глюкозамин, Аминоартрин, Эльбона, Юниум), Карнитина хлорид 20% раствор, Янтарную кислоту по 100-200 мг 2 раза в день, препараты магния (например, Магне В6), поливитаминно-минеральные комплексы (с кальцием, магнием, цинком, медью). Прием этих веществ направлен на нормализацию обмена веществ, укрепление соединительной ткани.
 Для лечения сердечно-сосудистых нарушений часто используют ?-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
Для лечения сердечно-сосудистых нарушений часто используют ?-адреноблокаторы (Пропранолол, Обзидан, Атенолол), блокаторы кальция (Нифедипин, Амлодипин, Лекоптин), ингибиторы ангиотензинпревращающего фермента (Лизиноприл, Периндоприл, Эналаприл), антиаритмические препараты (при нарушении ритма сердца). При развитии инфекционного эндокардита показаны антибиотики. После оперативных вмешательств может понадобиться антикоагулянтная терапия для снижения свертываемости крови (Фраксипарин, Клексан).
В целом подбор метода лечения и ассортимент применяемых лекарственных средств очень индивидуальны. Все зависит от спектра клинических симптомов и выраженности нарушений у конкретного больного.
Больным с синдромом Марфана показана лечебная физкультура, но в строго дозированном количестве, чтобы занятия приносили пользу сердечно-сосудистой и опорно-двигательной системам, а не вред. Нельзя заниматься контактными и игровыми видами спорта (баскетбол, футбол), рекомендовано плавание.
Синдром Марфана
Приветствую, дорогие читатели. Сегодня хотел бы поговорить про высоких людей с Синдромом Марфана, для которых высокий рост – это не просто особенность организма, а симптом серьезной болезни, требующей к себе постоянного внимания.
Синдром Марфана — заболевание наследственного типа, при котором поражается соединительная ткань с вовлечением в процесс скелетно-мышечной системы и глаз. Установлено, что причиной патологии является мутация гена фибриллина FBN1. Заболевание полиморфно — может протекать с разной выраженностью клинической картины, и характеризуется появлением все новых типов мутации в генах.

Синдром Марфана получил своё название от фамилии французского педиатра А. Марфана, который вперые представил описание 5-летней девочки Габриель с необычными, непрерывно прогрессирующими аномалиями скелета, и дал патологии своё имя.
Распространенность синдрома — 1 случай на 10000 человек. Риск рождения ребенка с синдромом Марфана повышается после достижения отцом возраста 35 лет и достигает 50% при наличии патологии у одного из родителей. Врожденная аномалия наследуется по аутосомно-доминантному типу. В ее основе лежит дефект важнейшего гена, отвечающего за синтез коллагена.
Во время внутриутробного развития происходит нарушение формирования волокон соединительной ткани, утеря ими прочности, в результате чего волокна не способны выдерживать естественные нагрузки. Поэтому наибольшие атипичные изменения претерпевают крупные сосуды, клапаны сердца, связки глаза, твердое небо, скелет и мышцы.
Без адекватной терапии продолжительность жизни людей с синдромом Марфана не более 40 лет. Терапия позволяет увеличить этот срок вдвое и более.
Классические симптомы Марфана
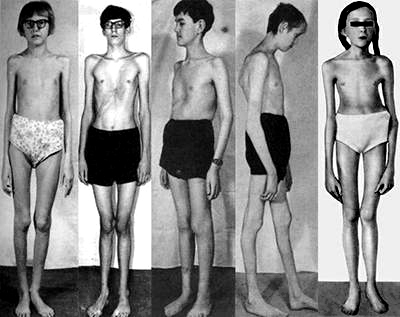
Классические проявления заболевания – это высокий рост, худощавое телосложение, искривление позвоночника или сколиоз, непропорционально по сравнению с туловищем длинные руки и ноги, длинные тонкие «паукообразные» пальцы, плохо развитые мышцы. Кожа у таких людей хрупкая, легко растягивается, но повышенную склонность к возникновению кровоизлияний. При обследовании сердечно-сосудистой системы обнаруживают расширение дуги аорты, различные варианты клапанных пороков сердца.
Глазные проявления
Близорукость является достаточно частым проявлением и наблюдается более чем у половины больных с синдромом Марфана, ее возникновение объясняется шаровидной формой хрусталика, изменением преломляющей силы роговицы глаза, а также растяжением самого глазного яблока.
Возникают также изменения в радужной оболочки, связанные с повышенной растяжимостью тканей. За счет этого возникают дефекты в радужке – так называемые колобомы, возможно закрытие угла передней камеры растянутой тканью радужки с повышением внутриглазного давления, то есть развитием глаукомы.
Из-за растяжения и слабости связок, на которых держится хрусталик глаза, так называемые связки Цинна, происходит их частичный или полный разрыв. Возникает либо подвывих хрусталика, когда из-за частичного разрыва связок хрусталик смещается, но все-таки держится на оставшихся связках. Или связки полностью отрываются, и хрусталик опускается в полость глаза, свободно меняя свое положение – возникает вывих хрусталика. Кроме того, помутнение хрусталика или катаракта развивается раньше и встречается более часто, чем у здоровых людей.
Глаукома возникает при нарушении оттока внутриглазной жидкости через угол передней камеры за счет закрытия измененной радужной оболочкой или закрытием путей оттока внутриглазной жидкости вывихнутым хрусталиком.
Избыточно растягивается также и сетчатка глаза, в результате чего повышается риск развития периферических хориоретинальных дистрофий – локальных истончений сетчатой оболочки глаз, которые могут приводить к возникновению отслойки сетчатки.
Наблюдается развитие косоглазия, появление которого также связано с повышенной растяжимостью тканей.
Методы коррекции и лечения
Близорукость можно корригировать при помощи очков или контактных линз.
При возникновении катаракты, либо развитии выраженного смещения хрусталика, которое снижает зрение или представляет опасность в плане развития глаукомы, проводят хирургическую операцию по удалению хрусталика и установки искусственной внутриглазной линзы.
При появлении дистрофий сетчатки с высоким риском развития отслойки проводят профилактическое лазерное лечение – наносят небольшие ожоги при помощи лазера, укрепляя сетчатку в местах ее истончения. Если же возникает отслойка сетчатки, то проводится обязательное хирургическое лечение.
Известные люди с синдромом Марфана
Синдром Марфана назван по имени педиатра, который наблюдал девочку с этим заболеванием на протяжении 20 лет. Имеется много интересных фактов о людях, имеющих характерные признаки патологии. Первая манекенщица (Лесли Хорнби – «Твигги»), которая была прототипом для всех чрезмерно худых моделей, болела синдромом Марфана. Наиболее известные личности, о которых есть подобные сведения:










- президент А. Линкольн,
- скрипач Н. Паганини,
- писатель Г. Х. Андерсен,
- композитор С. Рахманинов,
- олимпийский чемпион М. Фелпс.
Распространенность симптомокомплекса у многих одаренных людей дала возможность предположить, что он придает экстраординарные способности.
Пренатальное тестирование в диагностике синдрома Марфана
Как только мутация гена синдрома Марфана будет обнаружена, и пациент собирается стать родителем, есть возможность проверить своего будущего ребенка, чтобы выяснить, есть ли у него этот синдром. Вероятность того, что ребенок унаследует синдром, составляет 1 из 2 (50%). Для этого можно использовать 2 возможных теста: биопсию ворсин хориона (CVS) или амниоцентез.
Биопсия хориона. Пренатальное тестирование на синдром Марфана можно проводить примерно на 10–12 неделе беременности с использованием биопсии ворсин хориона. Процедура включает в себя взятие небольшого образца клеток из плаценты через вход в матку. Затем образец можно проверить на генетические условия.
Амниоцентез. Амниоцентез также можно использовать для диагностики синдрома Марфана. Анализ проводится на сроке от 16 до 18 недель беременности и включает взятие небольшого образца околоплодных вод для исследования. Хотя пренатальные тесты могут показать, есть ли у ребенка дефектный ген, вызывающий синдром Марфана, они не дадут никаких указаний на то, насколько серьезными будут их симптомы. Как правило, ребенок пострадает в той же степени, что и другие члены его семьи. Выраженность синдрома Марфана у родителя является показателем того, насколько тяжелым он будет у ребенка.
В некоторых случаях результаты биопсии хориона или амниоцентеза могут быть отрицательными, что позволяет предположить, что у младенца нет дефектного гена. Но у него может быть другая генетическая мутация, на которую не проводилось тестирование и которая все еще может вызывать синдром Марфана.
Лечение и профилактика осложнений
Специфической терапии заболевания не существует: изменить гены еще до рождения ребенка невозможно. Лечение только симптоматическое и зависит от тех изменений в организме, которые развиваются у больного синдромом Марфана. Некоторые осложнения патологии можно успешно корректировать, другие — устранять оперативным путем.
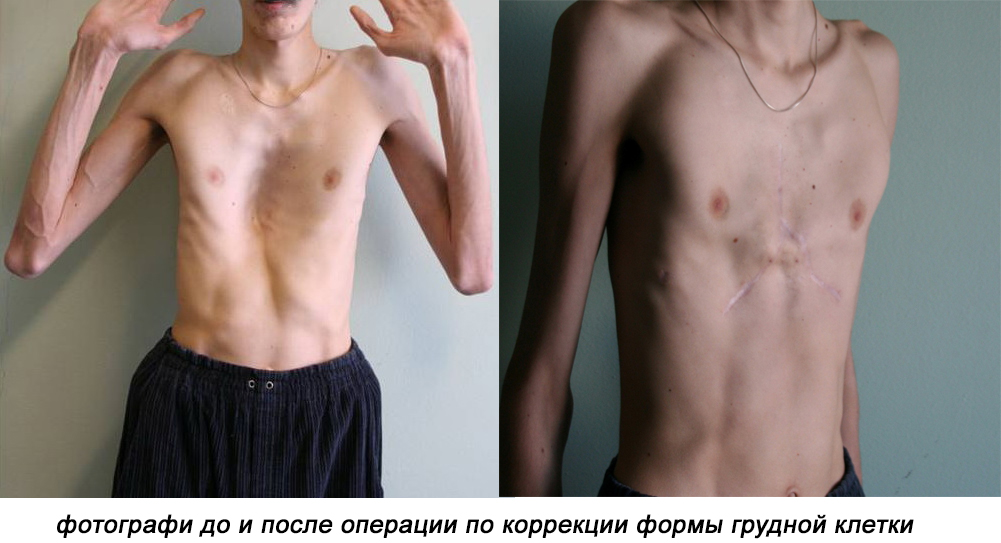
Пациент должен наблюдаться у группы специалистов — офтальмолога, невролога, кардиолога, ортопеда, хирурга. Основное направление терапии — поддержка функций сердца и сосудов.
Методы лечения: — прием препаратов (адреноблокаторы, антиаритмические лекарства, антикоагулянты и т.д.); — хирургия пороков сердца (дисфункции клапанов, расширения, расслоения легочной артерии), аорты, протезирование клапанного аппарата.
Нормализация зрения проводится при помощи коррекции миопии (ношение очков, линз), лечения катаракты, глаукомы, имплантации искусственного хрусталика.
При поражении суставов и позвоночника проводится оперативное лечение (протезирование, пластика суставов, устранение межпозвоночных грыж), выправление кифоза, сколиоза при помощи тракции, мануальной терапии. Из медикаментозных средств используются миорелаксанты, витамины группы В. Также применяется физиолечение, занятия ЛФК.
При поражении легких часто требуется хирургическое вмешательство (дренирование их полости).
Беременность больными синдромом Марфана должна строго планироваться и развиваться под контролем группы врачей, специализирующихся на лечении людей с подобными патологиями. Родоразрешение — только при помощи кесарева сечения. Еще до наступления беременности желательно обследоваться на предмет возможного прогрессирования расслойки аорты и, по возможности, провести операцию по замене части сосуда. Консультация генетика позволит рассчитать примерный риск по передаче заболевания по наследству.
Патологическая анатомия
При морфологическом исследовании — эластические волокна истончены, расположены неравномерно, местами хаотично; наблюдается расслоение средней оболочки крупных сосудов, разрыхление эндотелиального слоя, образование в эндотелиальном и субэндотелиальном слоях подушкообразных выступов в просвет сосуда (см. Аневризма расслаивающая). Эластический каркас аорты и легочного ствола развит слабо. Миокард с дистрофическими изменениями, вакуолизацией, местами с резким набуханием волокон. В костной ткани наблюдается разреженность костных балок и неравномерное отложение извести; нарушена структура хрящевой ткани за счет образования коллагеновых пучков, расслаивающих межуточное вещество.
Как проехать в медицинский центр
Кликните нужный район или метро
г. Москва, Сиреневый бульвар 32а. От метро Щелковская или Первомайская можно добраться пешком всего 10 минут более подробнее можете посмотреть в контактах.
- Телефон для справок:
- +7(495)500-93-90
Из района Измайлово добраться можно следующим образом: Маршрутка 1072 (до остановки метро Первомайская) далее 180 метров пешком.
- Телефон для справок:
- +7(495)500-93-90
Из Гальяново добраться можно следующим образом: Автобус номер 223, Троллейбус номер 23, Маршрутка 583 До остановки “Кинотеатр София”
- Телефон для справок:
- +7(495)500-93-90
От метро Черкизовская добраться можно следующим образом: автобус номер 230, до остановки “11 Парковая улица”
- Телефон для справок:
- +7(495)500-93-90
От метро Преображенская площадь добраться можно следующим образом: Маршрутка 1072 (до остановки метро Первомайская) далее 180 метров пешком.
- Телефон для справок:
- +7(495)500-93-90
От Поселка восточный можно добраться следующим образом: автобусы:283, 300,338, 349, 361 до остановки “11 Парковая улица”
маршрутки:1015, 102, 361, 362 до остановки “11 Парковая улица”
- Телефон для справок:
- +7(495)500-93-90
Лечение синдрома Марфана
Специфической терапии, направленной на устранение причины заболевания не существует. В настоящее время не разработаны методы влияния на наследственный аппарат клетки. Поэтому основная цель лечения синдрома Марфана – предотвращение прогрессирования заболевания, борьба с симптомами болезни:
патология сердца и сосудов.
Самым опасным проявлением недуга считается аневризма, расширение участка аорты. Коварство болезни заключается в непрерывном, длительном прогрессировании симптомов
Случается, что опасный симптом формируется к 18 годам, поэтому важно уделять достаточно внимания ежегодному обследованию и лечению сердечно-сосудистой системы
Грубые пороки развития сердца и сосудов, тяжёлые осложнения хронических болезней лечатся оперативно. Из лекарственных препаратов назначаются ингибиторы АПФ, блокаторы кальциевых каналов. Применение b-адреноблокаторов (пропанолола, атенолола) показано при расширении корня аорты, пролапсе клапанов, аритмиях.







Назначение препаратов и подбор необходимой дозировки должно проводиться врачом-кардиологом с учётом данных обследования ребёнка. Необоснованное назначение лекарственных средств может привести к ухудшению состояния ребёнка;

болезни опорно-двигательного аппарата.
Существую исследовании, указывающие на дефицит некоторых макроэлементов (кальций, цинк, кобальт, магний) и белков, необходимых для строительства соединительной ткани при синдроме Марфана. Поэтому для предотвращения прогрессирования патологии назначаются витаминно-минеральные комплексы, гиалуроновая кислота, викасол, колекальциферол. Оперативное лечение показано при грубых патологиях развития скелета;
заболевания органов зрения.
Исправление патологии зрения проводится с помощью подбора специальных очков, контактных линз, оперативного лечения катаракты и глаукомы, смещения хрусталика.
Опасным осложнением синдрома является отслойка сетчатки. Эта патологии возникает при активном занятии спортом у ребят с дефектом соединительной ткани. Повышенная физическая нагрузка, прыжки, травмы приводят к отделению тонкой сетчатой оболочки от сосудистой. Такое нарушение сопровождается резким снижением остроты зрения, которое не всегда является обратимым. Поэтому ребятам с генетическим синдромом следует избегать чересчур активных занятий, для таких детей хорошо подходит плавание в бассейне;
нарушенный обмен веществ.
Для улучшения метаболизма рекомендовано использовать в комплексном лечении аскорбиновую и янтарную кислоты, карнитин, препараты магния, токоферола ацетата. С целью нормализации обмена хрящевой ткани используются глюкозаминсульфат, хондроитинсульфат.
Существуют данные, указывающие на необходимость введения диеты с высоким содержанием магния для детей с синдромом Марфана. Этот элемент помогает справиться с повышенным содержанием катехоламинов в крови и способствует восстановлению дефектов кровеносных сосудов. Хорошо включать в ежедневный рацион орехи, какао, гречневую и ячневую каши, сухофрукты.