Как развивается заболевание
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов
Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного
Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.
Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
Симптомы синдрома Вильямса
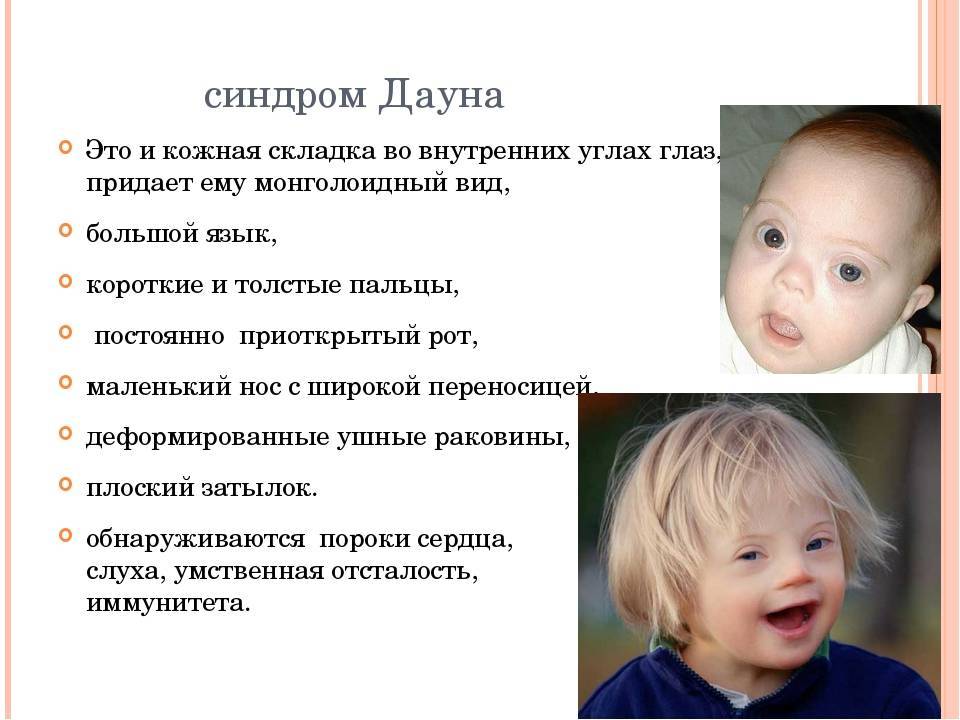
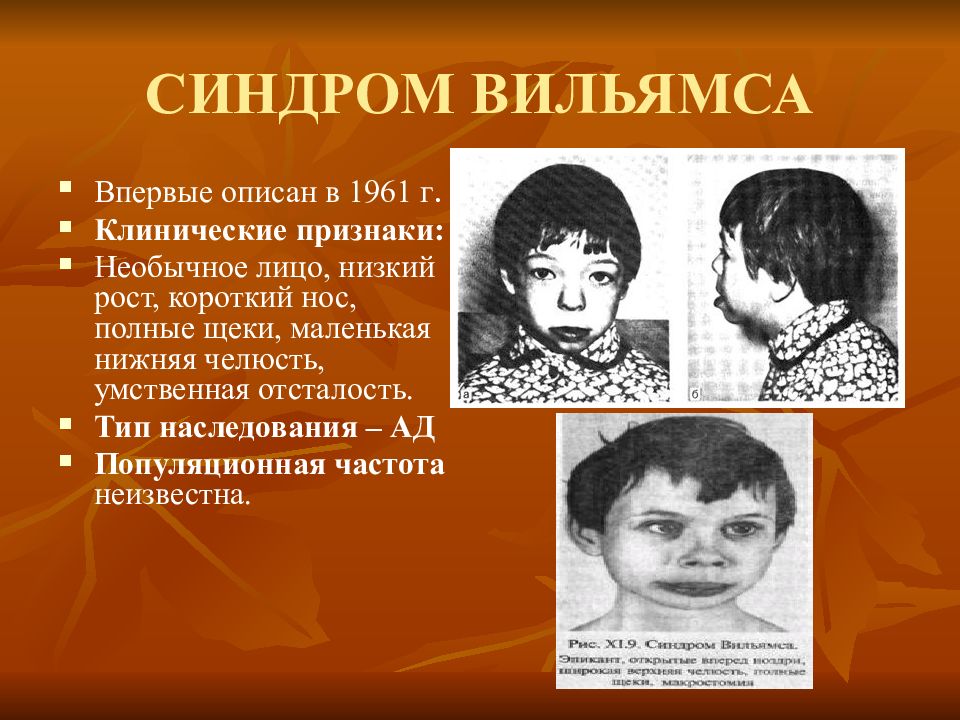
Проявления синдрома Вильямса иногда могут не определяться при рождении ребенка – в этот период заподозрить наличие заболевания можно лишь по сниженной массе тела, врожденному подвывиху бедра (наблюдается не у всех больных), порокам сердца. Все эти факторы достаточно неспецифичны, поэтому чаще всего патологию обнаруживают в старшем возрасте. Одним из самых заметных симптомов синдрома Вильямса является характерный внешний вид лица больных («лицо эльфа») – плоская переносица с округлым носом, увеличенный рот с приподнятыми вверх уголками, пухлые губы, полные щеки, небольшой заостренный подбородок. Из других изменений в области лица и головы часто отмечают низко посаженные уши, выступающий затылок, эпикантус. Для больных синдромом Вильямса характерен голубой цвет глаз с выраженным рисунком на радужной оболочке, голубоватый оттенок склер, отечность верхних и нижних век, нередко – косоглазие.
 Геморрой в 79% случаев убивает пациента
Геморрой в 79% случаев убивает пациента
Из патологических изменений при синдроме Вильямса чаще всего регистрируются различные пороки сердца, что в некоторых случаях может привести к раннему летальному исходу. В основном эти нарушения сводятся к недостаточности различных клапанов. Также у больных синдромом Вильямса чаще, чем у здоровых детей, возникают пупочные и паховые грыжи. Для этого заболевания характерна генерализованная мышечная гипотония, обуславливающая вторичные нарушения формирования скелета – Х-образные ноги, сколиоз и другие искривления позвоночного столба, плоскостопие, деформации грудной клетки. При осмотре стоматолога у больных синдромом Вильямса выявляются длинные, но редко расположенные зубы, которые довольно часто поражаются кариесом и легко разрушаются.
Дети, страдающие синдромом Вильямса, значительно отстают от сверстников, как в интеллектуальном, так и в физическом развитии. Они имеют пониженную массу тела при рождении, медленно набирают ее в дальнейшем, их рост останавливается намного раньше, поэтому проявлением этого заболевания также является низкорослость. Масса тела после подросткового периода может увеличиваться, имеется риск развития ожирения. В интеллектуальном плане больные синдромом Вильямса на всю жизнь сохраняют признаки имбецильности (умеренная умственная отсталость, средний уровень IQ 40-80). При этом они достаточно легко идут на контакт, у них, как правило, хорошо развита устная речь, при правильной коррекционной работе они способны выполнять простейшие бытовые поручения. В некоторых случаях при синдроме Вильямса может наблюдаться эмоциональная неустойчивость, тревожность, энурез.
Синдром Ангельмана: клинические симптомы
Впервые болезнь описал английский педиатр Г. Ангельман. Ее признаки бывает трудно определить до года. Явно выраженные симптомы проявляются с двухлетнего возраста. К этому времени уже заметны клинические проявления:
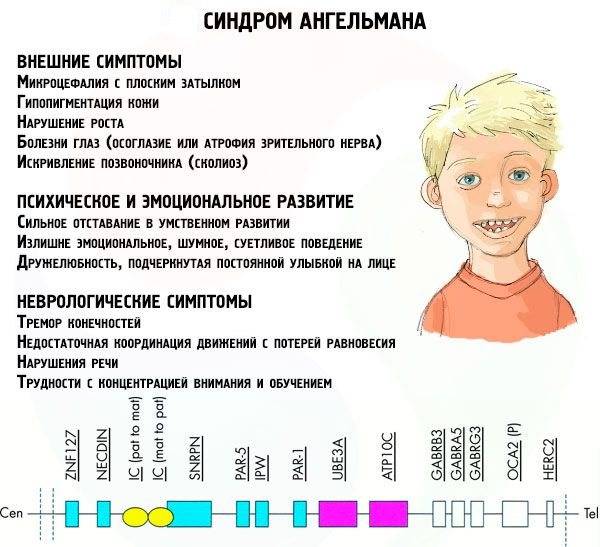
1. задержка психического развития – снижение когнитивных функций, умственная отсталость
2. речевые нарушения – ограниченная речь или ее отсутствие
3. психомоторные расстройства – хаотичные движения рук, тремор конечностей, атаксия (проблемы с координацией), специфическая походка
4. нарушения физического развития – черепно-лицевые аномалии: микроцефалия (объем черепа меньше нормы), редкие зубы, слюнотечение, уплощенный затылок, выступающая нижняя челюсть, открытый рот, высунутый язык. часто наблюдается сколиоз, мышечная дистония, косоглазие









5. эпилептическая активность, приступы судорог
6. особенности поведения (гиперактивность, смех без повода)
7. беспокойный сон.
Синдром Ангельмана у взрослых может быть менее выражен. С возрастом клинические проявления ослабевают – снижается гиперактивность, улучшается сон, уменьшается частота судорожных приступов. У них часто проявляются ожирение и сколиоз.
Внешние признаки при легкой форме болезни могут быть незаметными. Многие взрослые выглядят значительно моложе своих лет. Из-за выраженной задержки в развитии они в душе остаются детьми – могут быть импульсивными, чрезмерно обидчивыми, а иногда агрессивными.
История
Синдром Уильямса был впервые описан JCP Williams и его коллегами, которые написали в 1961 году о четырех пациентах с надклапанным стенозом аорты, умственной отсталостью и чертами лица, включая широкий лоб, большой подбородок, низко посаженные, «отвисшие» щеки, широко расставленные глаза. , и широко расставленный рот. Спустя год после этого отчета немецкий врач AJ Beuren описал трех новых пациентов с такой же картиной. Это привело к полному оригинальному названию синдрома, синдрому Вильямса-Бёрена, которое до сих пор используется в некоторых медицинских публикациях. С 1964 по 1975 год небольшие отчеты об исследованиях расширили медицинские знания о сердечно-сосудистых проблемах этого синдрома. Затем, в 1975 году, К. Джонс и Д. Смит провели крупномасштабный отчет о многочисленных пациентах с WS, в возрасте от младенчества до взрослого возраста, и описали поведенческие и наблюдаемые соматические симптомы более подробно, чем зарегистрировано ранее.
Лечение и профилактика
Влиять на генетический дефект невозможно, нарушения происходят на хромосомном уровне, поэтому больным назначают симптоматическое лечение и психологическую коррекцию.
Медикаментозная терапия может применяться при эпилептических приступах. Для смягчения симптомов заболевания и улучшения сна могут использоваться успокаивающие препараты.
Разрабатываются индивидуальные программы для каждого пациента – ЛФК, логопедические занятия, лечебный массаж, поведенческая терапия. Они дают возможность улучшить качество жизни человека. Также привлекают логопедов, дефектологов, специалистов по невербальным способам общения и поведенческой терапии.
В профилактических целях парам, планирующим зачатие ребенка, рекомендуется консультация генетика.
Общие сведения
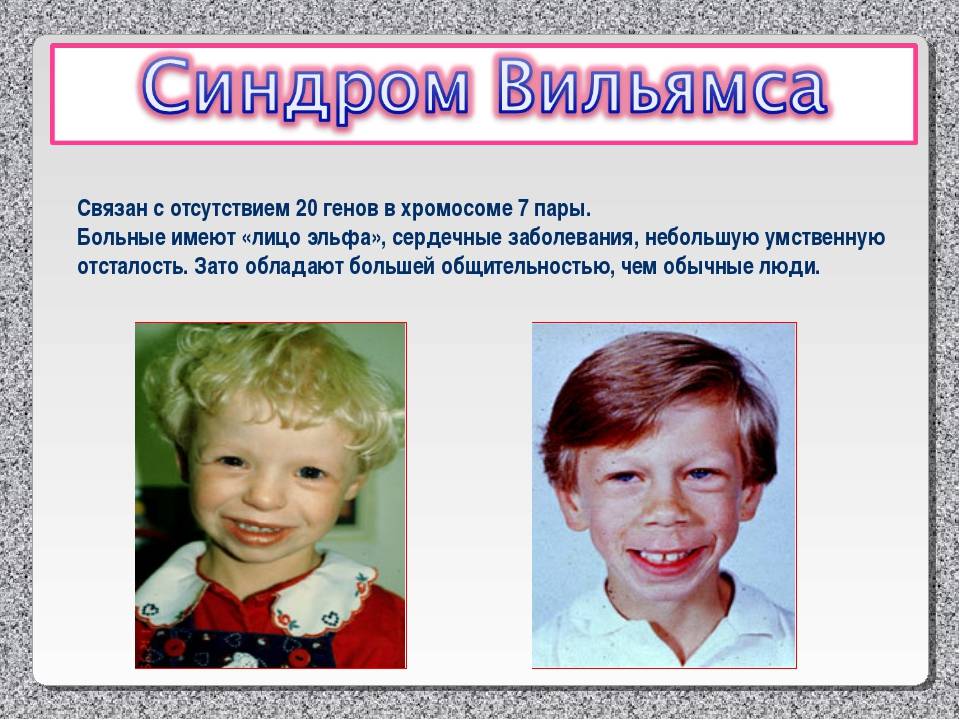
Синдром Вильямса (синдром Вильямса-Бойрена, синдром «лица эльфа») – генетическое заболевание, обусловленное отсутствием сразу нескольких генов, количество которых может колебаться от 25 до 29. С этим фактом связан довольно широкий спектр клинических проявлений состояния и различий в их выраженности.
Впервые данный синдром был описан в 1961 году кардиологом из Новой Зеландии Дж. Вильямсом – он выявил ряд фенотипически похожих детей со схожими врожденными нарушениями сердечно-сосудистой системы. В данный момент методами современной генетики удалось доказать наследственную природу синдрома Вильямса.
Синдром Вильямса
Причины возникновения синдрома Уильямса
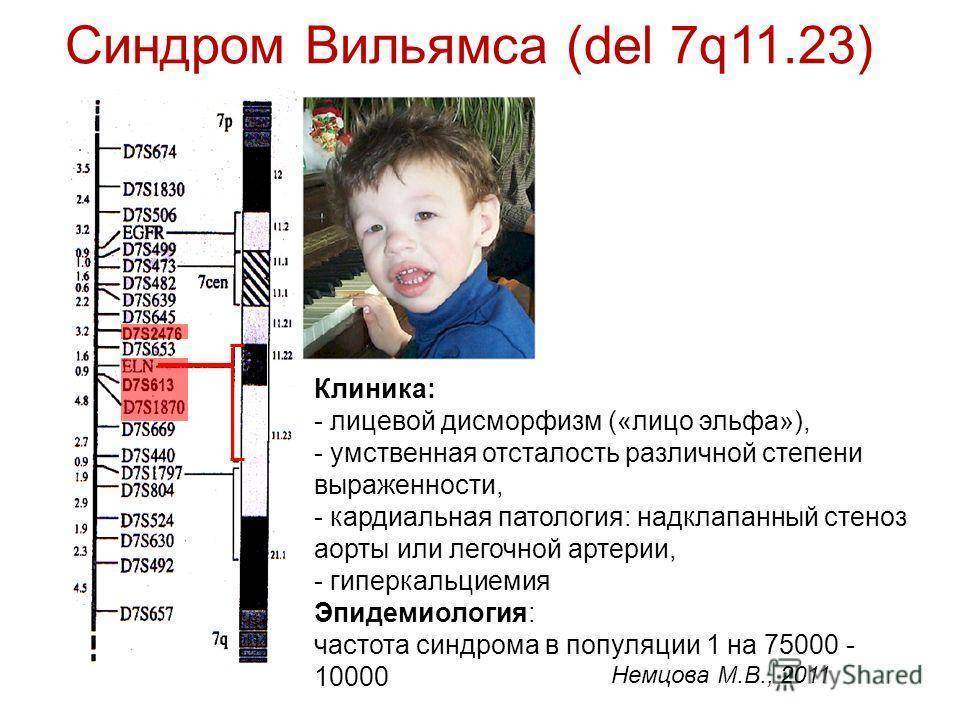
Ядро всех клеток молекул ДНК упаковано в нитеобразные строения, называемые хромосомами. В каждой клетке должно быть 46 хромосом. Каждая хромосома имеет короткое плечо (p) и длинное плечо (q). Если хромосом слишком много или слишком мало, или фрагмент хромосомы отсутствует, то речь идет о хромосомной аберрации. Синдром Уильямса — пример хромосомной аберрации, где отсутствует часть хромосомы 7. Другое название болезни — делеция 7q11.23 — это отсутствие части хромосомы (делеция) на длинном плече хромосомы 7.
Хромосома 7-одна из 23 четных хромосом человека. ДНК, составляющая хромосому 7, насчитывает более 158 миллионов пар оснований, что составляет около 5-5,5% генетического материала человека. Количество генов, имеющих свои локусы на хромосоме 7, оценивается в 1 000-1 400.
В недостающем фрагменте хромосомы содержится 26-28 генов, среди них очень важная роль отводится гену эластина, а также генам, ответственным за когнитивные функции. Отсутствие этих генов вызывает симптомы, возникающие при синдроме Уильямса. Наиболее изученный из них — ген эластина, представляющий собой белок, входящий в состав эластичных волокон, расположенных, среди прочего, в стенке кровеносных сосудов. Это объясняет нарушения со стороны сосудов и системы кровообращения, обнаруживаемые у большинства людей с этим генетическим синдромом.
Виды
Врачи выделяют врожденную и приобретенную энцефалопатию. Первая возникает на фоне неправильного течения беременности или родов и, зачастую, развивается еще во время пребывания плода в утробе матери. Ее признаки обнаруживаются сразу после родов или появляются в первые недели жизни. Диагностикой и лечением этого состояния занимаются неонатологи и педиатры.
Приобретенная энцефалопатия встречается уже во взрослом возрасте. Она подразделяется на несколько видов в зависимости от причины гибели нейронов:
- посттравматическая: возникает на фоне перенесенной черепно-мозговой травмы; зачастую, развивается уже через несколько лет после нее и нередко приводит к тяжелым расстройствам психики;
- токсическая: связана с острым или хроническим отравлением организма алкоголем, ядами, наркотическими препаратами, лекарственными средствами, солями тяжелых металлов и т.п.; нередко в рамках этого вида отдельно выделяют алкогольную энцефалопатию;
- метаболическая: связана с нарушением обмена веществ в организме; выделяют следующие подвиды патологии:
- печеночная: возникает при поражении печени или желчевыносящих путей;
- уремическая: связана с нарушением работы почек;
- диабетическая: является одним из частых осложнений сахарного диабета, возникает на фоне стойкого нарушения микроциркуляции и повышения вязкости крови;
- аноксическая: развивается после перенесенной клинической смерти и связана с кислородным голоданием головного мозга с последующим развитием «метаболической бури»;
- синдром Гайе-Вернике: энцефалопатия, вызванная дефицитом витамина В1;
- панкреатическая: является осложнением воспаления поджелудочной железы;
- гипогликемическая: возникает на фоне резкого снижения глюкозы крови;
- дисциркуляторная: связана с нарушением циркуляции крови в сосудах головного мозга; различают несколько форм патологии:
- атеросклеротическая: развивается из-за атеросклероза и утолщения стенок сосудов;
- гипертоническая: связана со стойким повышением артериального давления;
- венозная: возникает из-за нарушения венозного оттока крови.
В зависимости от скорости развития процесса выделяют энцефалопатию острую и хроническую. Первая может развиться в течение нескольких дней или часов, чаще возникает на фоне сильной интоксикации, травмы, инфекционного процесса. Хронический процесс может протекать годами и десятилетиями.
Генетические мутации повышают риск появления заболевания
Появление синдрома Ангельмана связано с наличием у родителей будущего ребенка различных хромосомных аномалий. Среди таких отклонений обычно называют:
- трисомию хромосом – присутствие одной или нескольких лишних хромосом в хромосомном наборе;
- инверсию – разворот одного из участков хромосомы на 180 градусов, при этом часть хромосомы пропущена, а гены располагаются в противоположном порядке;
- микроделецию, которая является результатом перестройки Y-хромосомы и обмена участками между хромосомами, наблюдается небольшое количество хромосом, а также может отсутствовать один из генов;
- делецию – нехватку одного из участков хромосомы;
- транслокацию – перенос или присоединение участка одной хромосомы к другой хромосоме;
- дупликацию – копирование части хромосом, результатом чего становится лишний генетический материал;
- кольцевую хромосому – на концах хромосомы отсутствует генетический материал, при этом новообразованные концы соединяются в виде кольца.
Генные мутации, которые могут вызвать развитие синдрома
Истоки заболевания
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.







В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, спустя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
Эпидемиология
Исторически считалось, что синдром Вильямса встречается примерно у одного из 20 000 живорождений, но более поздние эпидемиологические исследования показали, что этот показатель приближается к одному из каждых 7500 живорождений, что является значительно большей распространенностью. Поскольку все больше данных свидетельствует о том, что WS является более распространенным явлением, чем первоначально отмечалось (около 6% всех генетических случаев нарушения развития), исследователи начали выдвигать теории о недиагностировании этого синдрома в прошлом. Одна из теоретических причин увеличения эпидемиологических оценок заключается в том, что у значительной части людей с генетическими маркерами синдрома WS отсутствуют характерные черты лица или пониженный интеллект, который считается диагностическим признаком синдрома, и часто не сразу распознаются как имеющие синдром. .
«Он без общения умирает»
Порой Мише сложно отвечать на мои вопросы, и тогда он говорит односложно. Зато про любимые фильмы он рассказывает долго и подробно. Мише нравятся фильмы про романтику, нравится фильм про трех мушкетеров, который он смотрел около десяти раз. Из мушкетеров ему больше всего нравится его тезка. Еще отмечает фильм «Любовь — не картошка».
Больше всего Миша боится остаться один. Не только без мамы, но и без общения с ребятами. «Не люблю я быть один. Такой я человек, который не любит одиночество. Мне неуютно, мне не по себе. Приходишь домой — тишина, хочется с кем-то поговорить, — говорит Миша. — Не могу жить без людей, которые меня любят, уважают, ценят. Они мне важны, потому что я им важен. Если бы меня не было, я не знаю, как бы они жили без меня. Я им не чужой, я родной, любимый и не посторонний». У Миши мало друзей. Сейчас он общается с двумя парнями с синдромом Вильямса, а не по синдрому у него друзей нет. Раньше были школьные друзья, но общаться перестали.
Клиническая картина в подробностях
У разных больных могут проявляться разные симптомы. Эта разница зависит от степени и вида хромосомного отклонения.
Можно разделить признаки по частоте их проявления у детей, больных синдромом Ангельмана:
- Симптомы, которые проявляются у всех больных. К ним относят тяжелую функциональную задержку развития, отклонения в поведении (смех и улыбка, не имеющие причины, состояние счастья, повышенная возбудимость, пониженная концентрация внимания). Также у абсолютно всех пациентов наблюдается нарушение моторных функций, нарушение равновесия, тремор, преобладание невербальных навыков над словесными, нарушения речи.
- Симптомы, характерные для 80% пациентов. Задержка в росте, приводящая к диспропорции головы относительно других частей тела. Зачастую это приводит к развитию микроцефалии. У детей до трех лет часто проявляются эпилептические приступы, затем они становятся реже либо исчезают вовсе. Результаты электроэнцефалограммы часто аномальны, волны низкого уровня имеют повышенную амплитуду и временную динамику.
- Симптомы, возникающие у менее чем 80% больных. К таким проявлениям относят косоглазие, пониженную способность контролировать движения языка, проблемы с глотанием, гипопигментацию глаз и кожи, альбинизм, У многих пациентов наблюдается повышенная активность сухожильных рефлексов. В раннем детстве у многих возникают проблемы с питанием и сном. Среди проявлений также можно назвать слюнотечение, высовывание языка, постоянная жажда. К внешним признакам относят также наличие плоского затылка и гладких ладоней.
По мере взросления проявления синдрома изменяются. Реже наблюдаются расстройства сна, гиперактивность, судорожный синдром. Взрослые, имеющие синдром Ангельмана, выглядят моложе своих ровесников.
Половое созревание происходит немного позже, чем у индивидов, не имеющих данной патологии. Больные могут иметь собственных детей, однако высок риск передать заболевание потомству.
Многие взрослые пациенты страдают от неконтролируемого мочеиспускания. Часто встречается наличие сложностей с моторикой, что приводит к необходимости носить одежду без молний, пуговиц. Среди взрослых больных наблюдается проблема лишнего веса, поэтому необходимо контролировать соблюдение специальной диеты.
Дети хорошо воспринимают устную речь, и понимают содержание почти всех разговоров, однако редко отвечают. Часто они отказываются принимать участие в разговоре, и употребляют в речи несколько десятков слов.
Диагностика и своевременное выявление
Синдром куклы возможно диагностировать еще до рождения ребенка, путем проведения генетического исследования пятнадцатой хромосомы.
Диагностика может быть инвазивной и неинвазивной. Инвазивный метод исследования существует в медицине достаточно давно, однако является достаточно рискованным, так как необходимо проникновение в матку и забор околоплодной жидкости.
Неинвазивный метод предполагает анализ крови матери, при котором проводится анализ ДНК младенца. На основании данного исследования делаются выводы о присутствии или отсутствии определенных отклонений.
Диагностику также проводят новорожденным, у которых наблюдаются нарушения мышечного тонуса, отставание в развитии речи и моторики.
Необходимо следить за выражением лица младенца, его поведением, проявлением эмоций, движениями. Если у младенца имеются трудности со сгибанием конечностей, наблюдается тремор, хаотичные и резкие движения конечностей, то стоит обратиться за консультацией и помощью специалистов.
Зачастую диагноз синдром Ангельмана ставится в возрасте от 3 до 7 лет, когда можно заметить яркое проявление признаков данного заболевания. В зависимости от степени повреждений 15 хромосомы, проявления могут иметь различную тяжесть: некоторые пациенты имеют затруднения даже в речи, в то время как другие могут вести самостоятельную жизнь.
Из-за чего развивается патология
Синдром Вильямса («лица Эльфа») – это генетическое заболевание, связанное с нарушениями в хромосомном наборе плода. А именно с потерей участка (делецией) седьмой хромосомы. В утраченном так называемом «длинном плече хромосомы» содержится около 26 генов, что и связывают с характерными особенностями данного расстройства (хотя для большинства из них связь с имеющимися при болезни симптомами пока еще не установлена). Кроме того, что на поврежденном участке хромосомы имеются гены, отвечающие за то, как будет развиваться и функционировать головной мозг, там есть и гены, отвечающие за синтез белка эластина. А это приводит к тому, что у детей с описываемым синдромом возникают не только поведенческие, интеллектуальные и психологические проблемы, но и сосудистые аномалии, а также врожденные пороки сердца (стеноз аорты и легочной артерии, которые могут проявляться как по отдельности, так и вместе).
Нетипичная картина
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.
Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них
Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию
Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них
Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Первые годы жизни
Дети с синдромом Вильямса в первые два годы жизни ослаблены. У них плохой аппетит, постоянная жажда, часто возникает рвота и запоры, которые сменяются поносами. Часто наряду с этими симптомами отмечаются нарушения обмена веществ. Содержание в крови холестерина и кальция повышено.








Если ребенку поставлен диагноз «синдром Вильямса», то необходимо провести анализ на содержание в крови кальция, поскольку при этом заболевании концентрация кальция, как правило, повышена. Детям с нарушением кальциевого обмена назначают медикаментозное лечение и диету.
Соматическое состояние обычно улучшается к началу третьего года жизни, но при этом отставание в психомоторном развитии проявляется ярче.
Невзирая на отставание в развитии, общительность и адекватное поведение детей, их эмоциональность и стремление подражать взрослым, вселяют надежду на нормальное психическое развитие. Однако в дальнейшем эти надежды не оправдываются.